|
Step 1. Examine the alignment scores and statistics
|
|
Scores
|
|
Open a window with sample output.
The raw score "S" of the alignment is usually calculated by summing the scores for each letter-to-letter and letter-to-null position in the alignment.
Scores for each position of an alignment are derived from a substitution matrix, the most popular of these are the BLOSUM and PAM matrices.
Unlike the raw score, the bit score (shown to the right in the output window) accounts for the type of scoring system used, and is therefore more informative. The bit score is calculated from the raw score by normalizing with the statistical variables that define a given scoring system. Therefore, bit scores from different alignments, even those employing different scoring matrices can be compared.
The higher the score the better the alignment, but the significance of an alignment can not be deduced from the score alone. See Statistics below.
Positions at which a letter is paired with a null is called a gap. Gap scores are negative.
Since a single mutational event may cause the insertion or deletion of more than one residue, the presence of a gap is frequently ascribed more significance than the length of the gap. Hence the gap is penalized heavily, whereas a lesser penalty is ascribed to each subsequent residue in the gap.
There is no widely accepted theory for selecting gap costs. 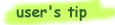 It is rarely necessary to change gap opening or extension values from the default. It is rarely necessary to change gap opening or extension values from the default.
|
|
Local alignments with no gaps are referred to as High Scoring Pairs (HSPs). The number of random HSP scores equal or greater than S is described by the Poisson distribution. This is the P value associated with the score S. Highly significant scores have P values close to zero.
For gapped alignments, the significance of a given alignment with score S is represented by the E (expect) value (shown in the right-most column in the output), the expected number of chance alignments with a score of S or better. This can be evaluated by looking at alignment scores generated using mock databases of random sequence of comparable length and composition.
The E value decreases exponentially as the Score (S) that is assigned to a match between two sequences increases.
The E value reflects the size of database and the scoring system in use.
At very low E values, the E and P values may converge.
A convenient way to create a significance threshold for reporting hits is to alter the E value. When the Expect value threshold is increased from the default value of 10, more hits can be reported.
|
|
Step 2. Examine the alignments
|
Descriptions
|
| |
The highest scoring alignments are described by one line summaries called "descriptions".
The description lines are sorted by increasing E value, thus the most signficant alignments (lowest E values) are at the top.
The description consists of four columns (from the left): (1) identifier for the database sequence; (2) brief description of the sequence; (3) the (bit) score of the highest-scoring HSP found for each database sequence; (4) the E value.
The identifer is linked to the full GenBank entry.
Clicking on the score in a given description line will take the user to the corresponding sequence alignment. The alignment can also be reached by scrolling down the output pages.
|
|
The colored bars in the graphic summarize the BLAST results (consult figure).
At the top is a linear map of the query. Each bar drawn below the map represents a protein (or protein fragment) that matches the query sequence. The position of each bar relative to the linear map of the query allows the user to see instantly the extent to which the database matches align with a single or multiple regions of the query.
The most similar hits are shown at the top in red. Pink, green, blue and black bars follow, representing proteins in decreasing order of similarity. Hatched areas (when present) correspond to the non-similar sequence between two or more distinct regions of similarity found within the same database entry.
Moving the mouse over the bars will display the name of the matching protein found in the textbox above.
|
| |
Alignments can be represented in a variety of formats selected by the user either before or after the query is submitted.
The default format is the "pairwise alignment" in which the aligned positions of the query and the database match (the subject) are arranged with one vertical space between them. In protein alignments, identical residues are listed in the middle. Conserved residues are represented by plus signs. In DNA alignments, vertical lines connect identical residues.
Gaps are represented as dashes within the query or subject sequence.
Due to filtering, an amino acid query sequence may contain X's in place of low complexity sequences. (N's in a nucleic acid query). This accounts for a decrease in identity and increase in E value than would otherwise be seen in a match of a query against the identical or other highly related sequences in the database.
More than one alignment per database entry may be listed among the HSPs.
|
|
Step 3. Review details of the search process
|
BLAST search parameters specified in the query
|
|
Which database was searched (#1)
Which matrix was used during the search (#5)
Gap creation and gap extension costs (#6)
|
|
Date on which the query database was built (#2)
Size of the database at that time (#3)
|
|
Statistics of the BLAST search
|
|
Values for lambda, K, and H calculated from the results of the search (ungapped, gapped) (#4)
BLAST statistics (#7)
|
